Navigation auf uzh.ch
Navigation auf uzh.ch
Ever since the prion gene was discovered in 1985, its role and biological impact on the neurons has remained a mystery. “Finally, we can ascribe a clear-cut function to prion proteins and reveal that, combined with particular receptor, they are responsible for the long-term integrity of the nerves,” says Professor Adriano Aguzzi from the Neuropathological Institute at the University of Zurich and University Hospital Zurich. The present study therefore clears up a question that researchers have been puzzling over for 30 years, but ultimately went unanswered.
Prions are dangerous pathogens that trigger fatal brain degeneration in humans and animals. In the 1990s, they were responsible for the BSE epidemic more commonly known as mad cow disease. In humans, they cause Creutzfeldt-Jakob disease and other neurological disorders that are fatal and untreatable. Meanwhile, we know that infectious prions consist of a defectively folded form of a normal prion protein called PrPC located in the neuron membrane. The infectious prions multiply by kidnapping PrPC and converting it into other infectious prions.
For a long time, it remained unclear why we humans – like most other organisms – have a protein in our neurons that does not perform any obvious function, yet can be extremely dangerous. Aguzzi has spent decades researching this issue and examining the theory that animals without the PrPC gene are resistant to prion diseases. But what are the repercussions for the organism if the prion protein is deactivated?
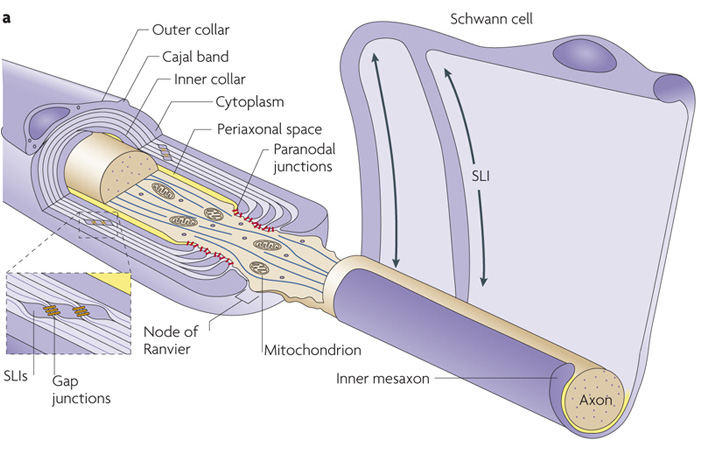
A few years ago, Aguzzi and his team discovered that mice without the PrPC gene suffer from a chronic disease of the peripheral nervous system. The reason: The so-called Schwann cells around the sensitive nerve fibers no longer form an insulating layer to protect the nerves. Due to this insulating myelin deficit, the peripheral nerves become diseased, potentially resulting in motoric disorders in the motion tract and paralysis.
The researchers have now gone one step further in the lab: In a new study, Alexander Küffer and Asvin Lakkaraju clarify exactly why the peripheral nerves become damaged in the absence of the prion protein PrPC. They discovered how the PrPC produced by the neurons docks onto the Schwann cells: namely via a receptor called Gpr126. If the prion protein and the receptor work together, a particular messenger substance (cAMP) which regulates the chemical interaction in the cells and is essential for the integrity of the nerve’s protective sheath increases. Gpr126 belongs to the large family of “G-protein-coupled receptors”, which are involved in many physiological processes and diseases.
This discovery solves a key question that has long puzzled neuroscientists and points towards future applications in hospitals. “If you want to deactivate the prion protein PrPC fully for potential Creutzfeld-Jakob disease treatments, you need to know the potential side effects on the nerves in the future,” explains Aguzzi. Moreover, the present results on the effect of PrPC at molecular level could yield a new approach for peripheral neuropathy. Currently, there are only extremely limited therapeutic options for these chronic debilitating diseases of the nervous system.
Alexander Küffer, Asvin K. K. Lakkaraju, Amit Mogha, Sarah C. Petersen, Kristina Airich, Cédric Doucerain, Rajlakshmi Marpakwar, Pamela Bakirci, Assunta Senatore, Arnaud Monnard, Carmen Schiavi, Mario Nuvolone, Bianka Grosshans, Simone Hornemann, Frederic Bassilana, Kelly R. Monk & Adriano Aguzzi. The prion protein is an agonistic ligand of the G-protein-coupled receptor Gpr1/Adgrg6. Nature, 8 August 2016. doi:10.1038/nature19312